Cross-Reactive-Epitope-Search-using-Structural-Properties-of-proteins (CRESSP)
A program to find cross-reactive epitopes with structural information from known protein structures.
Introduction
Our new pipeline, called Cross-ReactiveEpitope-Search-using-Structural-Properties-of-proteins (CRESSP), use structural information from RCSB-PDB database to search potential cross-reactive B-cell epitopes of human and pathogen proteins.
First, protein sequences of interest (provided by user) are searched with either BLASTP alone or in combination with HMMER3 with an HMM profile database built from ~5000 Pan-Proteomes from the UniProt database.
Second, using pre-computed experimental and predicted relative-surface-availability (RSA) values of human protein residues, the alignment between human and pathogen proteins are further analyzed to identify potential cross-reactive B-cell epitopes. The RSA-weighted BLOSUM62 scores are calculated with an array of sliding windows (provided by user) to estimate possible cross-reactivity between two proteins.
Lastly, the output file from our pipeline can be visualized interactively with web-browser-based application (similar to the interactive interface in our web-application for SARS-CoV-2, http://ahs2202.github.io/3M/)
Currently, we are additionally implementing neural-network-based (bi-directional stacked RNN) surface-availability prediction module in our tool to efficiently predict RSA values of any protein sequences of interests so that CRESSP can predict B-cell cross-reactivity between any proteome of interest, including proteins from metagenome-assembled genomes.
Installation
CRESSP requires BLASTP from BLAST+ (v2.10.1+) and HMMER3, which can be installed through conda with the following commands:
conda install -c bioconda blast
conda install -c bioconda hmmer
BLAST+ binaries can be also downloaded from here.
CRESSP can be installed from PyPI (https://pypi.org/project/cressp/)
pip install cressp
CRESSP uses TensorFlow (>2.3.0) for prediction of relative surface area (RSA) and secondary structure classification. If CUDA-enabled GPU is available, CRESSP will automatically use GPU for prediction of structural properties of input proteins.
For T-cell cross-reactive epitope prediction, CRESSP uses MHCflurry 2 to predict MHC binding affinity values of aligned peptides. Before using CRESSP, MHCflurry 2 models and datasets should be downloaded by executing the following command:
mhcflurry-downloads fetch
Usage
command-line interface
usage: cressp [-h] [-t DIR_FILE_PROTEIN_TARGET] [-q DIR_FILE_PROTEIN_QUERY] [-o DIR_FOLDER_OUTPUT] [-c CPU] [-w WINDOW_SIZE] [-e FLOAT_THRES_E_VALUE] [-H] [-d DIR_FILE_QUERY_HMMDB] [-R] [-Q]
[-s FLOAT_THRES_AVG_SCORE_BLOSUM_WEIGHTED] [-S FLOAT_THRES_AVG_SCORE_BLOSUM] [-C FLOAT_THRES_RSA_CORRELATION]
Cross-Reactive-Epitope-Search-using-Structural-Properties-of-proteins (cressp): a program to find cross-reactive epitopes with structural information from known protein structures.
optional arguments:
-h, --help show this help message and exit
-t DIR_FILE_PROTEIN_TARGET, --dir_file_protein_target DIR_FILE_PROTEIN_TARGET
(Required) an input FASTA file containing target protein sequences.
-q DIR_FILE_PROTEIN_QUERY, --dir_file_protein_query DIR_FILE_PROTEIN_QUERY
(Default: UniProt human proteins) an input FASTA file containing query protein sequences.
-o DIR_FOLDER_OUTPUT, --dir_folder_output DIR_FOLDER_OUTPUT
(Default: a subdirectory 'cressp_out/' of the current directory) an output directory
-c CPU, --cpu CPU (Default: 1) Number of logical CPUs (threads) to use in the current compute node.
-w WINDOW_SIZE, --window_size WINDOW_SIZE
(Default: 30) list of window sizes separated by comma. Example: 15,30,45
-e FLOAT_THRES_E_VALUE, --float_thres_e_value FLOAT_THRES_E_VALUE
(Default: 1e-20) threshold for the global alignment e-value in a scientific notation Example: 1e-3
-H, --flag_use_HMM_search
(Default: False) Set this flag to perform HMM search in addition to BLASTP search. HMM profile search is performed with HMMER3. The search usually takes several hours for
metagenome-assembled genomes
-d DIR_FILE_QUERY_HMMDB, --dir_file_query_hmmdb DIR_FILE_QUERY_HMMDB
(Default: a HMM profile database of 1012 human proteins searched against UniProt Pan Proteomes. These proteins consist of experimentally validated human autoantigens) a file
containing HMM DB of query proteins aligned against pan-proteomes
-R, --flag_use_rcsb_pdb_only
(Default: False) When calculating consensus structural properties of input proteins, do not use homology-based modeled structures from SWISS-MODEL repositories and only use
experimental protein structures from RCSB PDB
-Q, --flag_only_use_structural_properties_of_query_proteins
(Default: False) Only use estimated structural properties of the query proteins when predicting cross-reactivity between target and query proteins (skip the estimation of
structural properties of target proteins). When 'dir_file_protein_query' == 'human' (default value), it will significantly reduce computation time by skipping estimation and
prediction steps of structural properties of target and query proteins.
-s FLOAT_THRES_AVG_SCORE_BLOSUM_WEIGHTED, --float_thres_avg_score_blosum_weighted FLOAT_THRES_AVG_SCORE_BLOSUM_WEIGHTED
(Default: 0.15) threshold for average weighted BLOSOM62 alignment score for filtering predicted cross-reactive epitopes
-S FLOAT_THRES_AVG_SCORE_BLOSUM, --float_thres_avg_score_blosum FLOAT_THRES_AVG_SCORE_BLOSUM
(Default: 0) threshold for average BLOSOM62 alignment score for filtering predicted cross-reactive epitopes
-C FLOAT_THRES_RSA_CORRELATION, --float_thres_rsa_correlation FLOAT_THRES_RSA_CORRELATION
(Default: 0) threshold for correlation coefficient of Relative Surface Area (RSA) values for filtering predicted cross-reactive epitopes
usage as a python package
To run CRESSP from an interactive Python environment, the following code can be used.
import cressp
cressp.cressp( dir_file_protein_target = "UP000464024_2697049.fasta", n_threads = 2, flag_use_HMM_search = True, l_window_size = [ 15, 30 ], float_thres_e_value = 1e-2 )
Alternatively, to run only a part of CRESSP to estimate structural properties of protein sequences of interest, the following code can be used.
import cressp
import cressp.structural_property_estimation
cressp.structural_property_estimation.Estimate_structural_property( "UP000464024_2697049.fasta", n_threads = 2, dir_folder_pipeline = "output/" )
dict_sp = cressp.Parse_Structural_Properties( "output/UP000464024_2697049.tsv.gz" )
visualization of analysis results with CRESSP-web-viewer
Open CRESSP Web Viewer CRESSP_Web_Viewer.html in the output directory with a web browser.
CRESSP Web Viewer can be also found here.
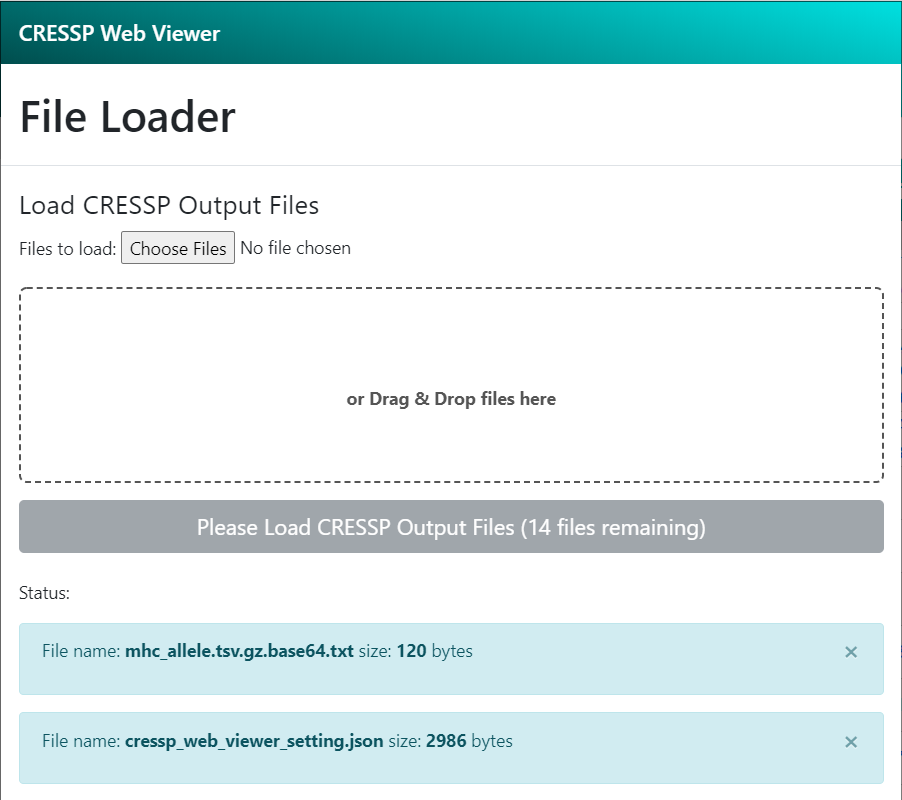
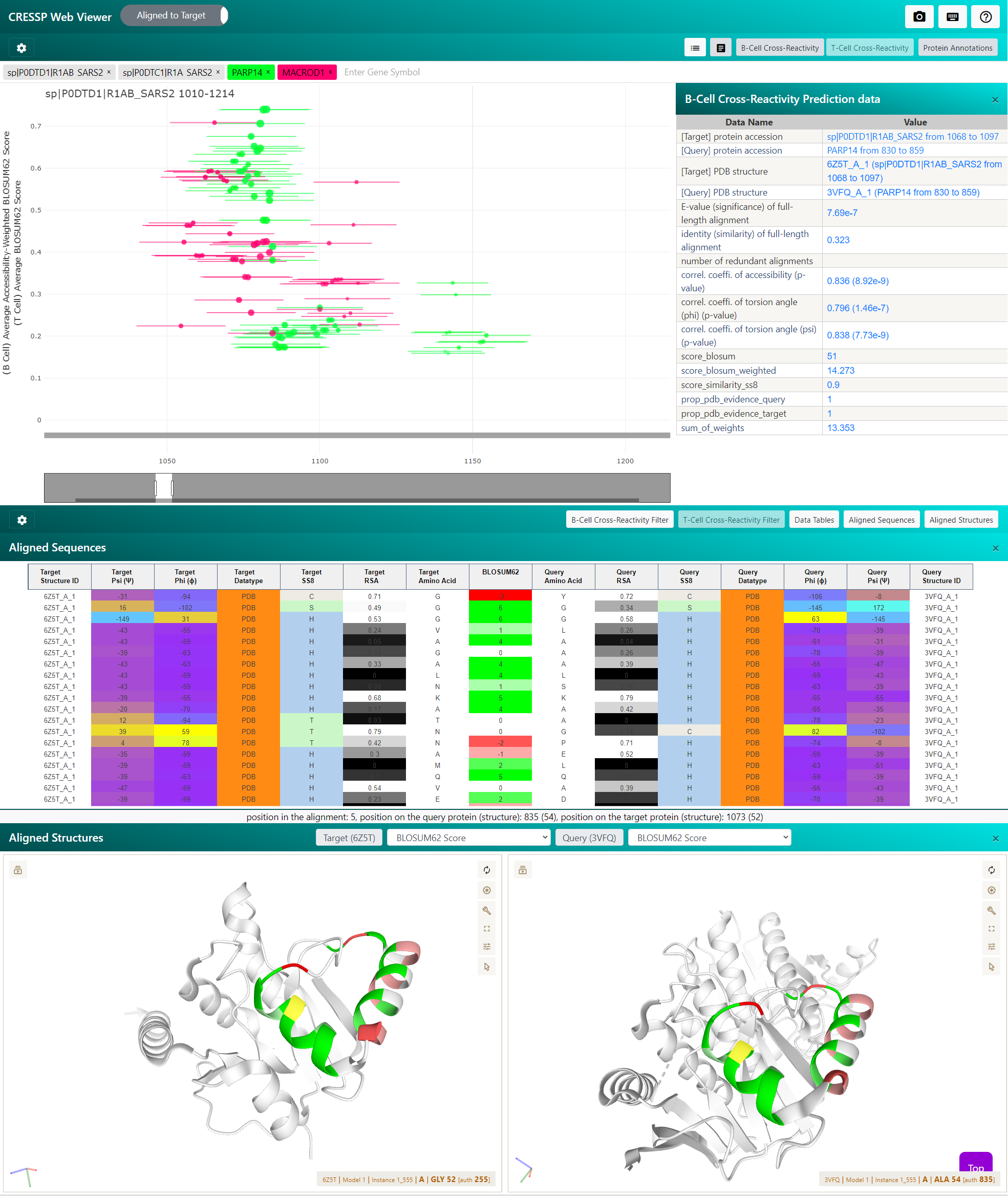
Then, load (drag & drop) CRESSP output files in the web_application/ subdirectory of the output directory. Once all necessary files has been loaded, the text on the grey button will change into 'Start Analysis'. Click the button to explore CRESSP analysis results. (Below is an example screen of CRESSP Web Viewer). Example CRESSP output files can be found at here.
Tutorial
cressp -t UP000464024_2697049.fasta.gz --flag_use_HMM_search --float_thres_e_value 5e-2 --cpu 2
Running CRESSP with the above command will do the following tasks:
- Search SARS-CoV-2 protein sequences with human protein sequences with BLASTP and HMMER3, and
- Align SARS-CoV-2 protein sequences against known or predicted protein structures from the RCSB-PDB database and the SWISS-MODEL Repository.
- Predict structural properties (surface accessibility and secondary structure) of protein residues not covered by the protein structure databases using Deep Neural Network (DNN) models trained on the protein structure databases.
- Calculate similarity scores based on estimated structural properties of SARS-CoV-2 proteins and (pre-computed) structural properties of human proteins