REvolutionH-tl is a powerful Python tool designed for evolutionary analysis tasks. It provides a comprehensive set of features for orthogroup analysis, gene tree reconstruction, species tree reconstruction, reconciliation of gene and species trees, and visualization of gene content evolution.
[1] Ramírez-Rafael J. A., Korchmaros A., Aviña-Padilla K., López-Sánchez A., Martinez-Medina G., Hernández-Álvarez A., Hellmuth M., Stadler P. F., and Hernandez-Rosales M. (2025) REvolutionH-tl: A Fast and Robust Tool for Decoding Evolutionary Gene Histories. Submitted.
[2] Ramírez-Rafael J. A., Korchmaros A., Aviña-Padilla K., López-Sánchez A., España-Tinajero A. A., Hellmuth M., Stadler P. F., and Hernandez-Rosales M. (2024) REvolutionH-tl: Reconstruction of Evolutionary Histories tool. In Comparative Genomics: 21st International Conference, RECOMB-CG 2024, Boston, MA, USA, April 27–28, 2024, Proceedings. Springer-Verlag, Berlin, Heidelberg, 89–109. https://doi.org/10.1007/978-3-031-58072-7_5
This guide will walk you through the installation, usage, and key functionalities of REvolutionH-tl.

Installation
To get started with REvolutionH-tl, you can easily install it using pip:
pip install revolutionhtl
Extra requirements
To perform sequence alignments, you'll also need to install Diamond or BLAST. You can download the Diamond executable from here or use the command line as follows:
wget http://github.com/bbuchfink/diamond/releases/download/v2.1.8/diamond-linux64.tar.gz
tar xzf diamond-linux64.tar.gz
To install BLAST follow these instructions: Unix / Windows. Or use the command line:
apt-get install ncbi-blast+
To generate the figure change in gene content, you'll need to install the following packages in your conda environment.
conda install -c conda-forge r-base=4.4.0
conda install -c conda-forge poppler imagemagick rust ffmpeg libxml2 librsvg tesseract
Next, you'll need to install the required R packages. To do this, follow these steps.
Download the package installation script:
wget "https://github.com/gabiiMM/RevolutionH-tl_Plot_Extension/raw/main/R_packages.R" .
Run the R package installation script:
Rscript R_packages.R
Synopsis
REvolutionH-tl is a command-line tool, and it can be used with the following syntax:
python -m revolutionhtl <arguments>
In addition, use the commands for visualization of results:
python -m revolutionhtl.plot_summary <arguments>
python -m revolutionhtl.plot_reconciliation <orthogroup IDs> <arguments>
For examples click here.
Let's delve into the steps of the program, the required arguments, and output files:
Input/Output Overview
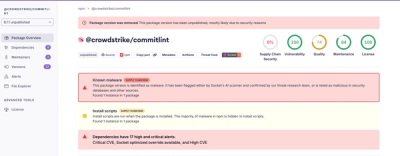
The REvolutionH-tl methodology is divided into 6 steps (see figure above). You can run all of them in a row by providing a directory containing fasta files (use the argument -F <directory>). Furthermore, if you want to run a specific step using precomputed files, you can use the argument -steps <step list> together with the arguments required for such a step.
When several steps of REvolutionH-tl are executed in a row and the tool has to decide between manually specified files and files produced at a previous step, the tool prioritizes the second option. For example, when running steps 3 and 6, step 3 generates gene trees that will be used as input for step 6 even if the user specifies a different set of gene trees. To avoid this behavior, run step 6 independently.
Workflow
-
Alignment Hits Computation. First, REvolutionH-tl runs a sequence aligner to obtain alignment hits and statistics like bit-score and e-value.
Required argument: -F <directory containing fasta files>,
Output: directory tl_project.alignment_all_vs_all/.
-
Best Hit, Orthogroup Selection, and Distance estimation. Best hits are the putative closest evolutionary-related genes across species, and orthogroups are a collection of genes sharing a common ancestor. Gen-to-gene distances are estimated using the scoredist approximation.
Input argument: -alignment_h <directory containing alignment hit files> (generated at step 1).
Optional: -OG <.tsv file containing orthogroups>.
Output files: tl_project.best_hits.tsv, tl_project.orthogroups.tsv, tl_project.distances.tsv.
-
Gene Tree Reconstruction, Best Matches, and Orthology Assignment. Gene trees are reconstructed from the hits, and the inner nodes of the trees are labeled as 'speciation' or 'duplication' events. Best matches are the closest evolutionary-related genes across species based on gene tree topology. Two genes are orthologous if they were conserved after a speciation process.
Required argument: -best_h <.tsv file containing best hits> (generated at step 2).
Output files: tl_project.gene_trees.tsv, tl_project.orthologs.tsv, tl_project.best_matches.tsv.
-
Duplication Polytomie Resolution. Duplication-labeled polytomies on the gene trees are resolved using a NJ approach. The resolved nodes correspond to duplication nodes contained in a single branch of the species tree.
Required arguments:
-T <.tsv file containing gene trees> (generated at step 3).
-D <.tsv file containig gene-to-gene distances> (generated at step 2),
Output file: tl_project.resolved_trees.tsv.
-
Species tree reconstruction. Species trees are obtained as a consensus of the speciation events in the gene trees.
Required argument: -T <.tsv file containing gene trees> (generated at steps 3 and 4).
Output file: tl_project.species_tree.nhx.
-
Tree reconciliation. Tree reconciliation depicts the evolution of genes across existing and ancestral species, it is represented as a gene tree embedded into a species tree.
Required arguments:
-T <.tsv file containing gene trees> (generated at steps 3 and 4).
-S <single-line file containing species tree> (generated at step 5).
Output files: tl_project.reconcilied_trees.tsv, tl_project.corrected_trees.tsv, tl_project.labeled_species_tree.nhx.
You can speed up the analysis by using the multithreading arguments -t <nuber of threads> and -j <number of jobs>.
To see the complete list of arguments for the main workflow please use python -m revolutionhtl -h, alternatively, you can check the list below.
Input data (Click to expand)
-h Show the full help message and exit.
-steps List of steps to run (default: 1 2 3 4 5 6).
-F [str | Input for step 1] Directory containing fasta files.
-alignment_h [str | Input for steps 2 and 4] Directory containing alignment hits.
-OG [str | Optional input for step 2] File specifying orthogroups for the best hit graph construction.
-best_h [str | Input for step 3] .tsv file containing best hits.
-T [str | Input for steps 4, 5, and 6] .tsv file containing gene trees in nhx format.
-D [str | Input for step 4] .tsv file containing gene-to-gene distances.
-S [str | Input for step 6] .nhx file containing a species tree.
File names (Click to expand)
-o [str | Default: tl_project] Prefix for output files.
-fext [str | Prameter for steps 1 and 2 | Default: .fa] Extesion for fasta files.
-no_singletons [flag | Prameter for step 2] Use to avoid singletons identification, i.e. genes that are in fasta files but are not assigned to an orthogroup.
-og [str | Default: OG] Column specifying orthogroup ID in input and output .tsv files.
-Nm [int | Default: 2000] Maximum number of genes in an orthogroup, bigger orthogroups are split. If 0, no orthogroup is split.
-S_attr [str | Default: ;] Attribute delimiter in the input .nhx file (Input of step 5).
Algorithm parameters (Click to expand)
-aligner [str | Parameter for step 1 | Default: diamond] Command or path to the program for computation of alignment hits. Supported: diamond, blastn, blastp.
-v [flag | Parameter for step 1 ] Use to display diamond messages.
-e [float | Parameter for step 1 | Default: 1e-05] Maximum evalue required to consider significant an alignment hit.
-m_command [str | Parameter for step 1 | Default: makeblastdb] BLAST command or path to the BLAST program for database creation.
-id [float | Parameter for step 1 | Default: 25] Minimum percentage of identity required to report an alignment hit.
-cov [float | Parameter for step 1 | Default: 50] Minimum percentage of query coverture required to report an alignment hit.
-k_hits [int | Parameter for step 1 | Default: 100] Maximum number of alignment hits per gene against a fixed species.
-bh_heuristic [str | Parameter for step 2 | Default: target] Indicates how to normalize bit-score. Normalize by sequence length: query, target, alignment, smallest. No normalization: row.
-f [float | Parameter for step 2 | Defualt: 0.95] Number between 0 and 1. Defines the adaptative threshold for best-hit selection as: f*max_bit_score.
-bmg_h [str | Parameter for step 3 | Default: Louvain] Comunity detection method for MaxConsistentTriples heuristic. Options: Mincut, BPMF, Karger, Greedy, Gradient_Walk, Louvain, Louvain_Obj.
-no_binary_R [flag | Parameter for step 3 ] Use to avoid the usage of binary triples from the best-hit graph.
-f_bT [flag | Parameter for step 3 ] Use to force gene trees to be binary.
-T_no_db [flag | Parameter for step 3 ] Use to avoid running build twice in the MaxConsistentTriples heuristic.
-stree_h [str | Parameter for step 5 | Default: louvain_weight] Comunity detection method for MaxConsistentTriples heuristic. Options: naive, louvain, mincut, louvain_weight.
-streeh_repeats [int | Parameter for step 5 | Default: 4] Specifies how many times run the MaxConsistentTriples heuristic.
-streeh_b [flag | Parameter for step 5] Use to force species tree to be binary.
-streeh_ndb [flag | Parameter for step 5] Use to avoid running build twice in the MaxConsistentTriples heuristic.
-n_edit_T [flag | Prameter for step 6] Use to avoid editing of inconsistent gene trees.
Visualization
-
Summary plot This command automatically searches in the current directory for the output files of revolutionhtl: tl_project.labeled_species_tree.nhx, tl_project.reconcilied_trees.tsv, tl_project.orthogroups, and tl_project.orthologs.
You can change the prefix of input files (tl_project.) using the argument -files_prefix <custom prefix>, you can also change the input/output path using -files_path <custom path>.
Furthermore, you can specify input file names using -S <species tree>, -R <reconcilied trees> -OG <orthogroups>, and -OR orthologs.
Output figures: tl_project.reconciliation_tree.pdf, tl_project.change_in_gene_content.pdf
Optional arguments: --OGs_mask <list of orthogroups for visualization>, --size <length in inches>, and --percentage_upsetplot <percentage (int) of rows shown in upsetplot>.
-
Reconciliation plot This command automatically searches in the current directory for the output files of revolutionhtl: tl_project.labeled_species_tree.nhx, and tl_project.reconcilied_trees.tsv.
Additionally, you have to provide a list of orthogroup identifiers to be displayed. These IDs have to be present in the column 'OG' of the file tl_project.reconcilied_trees.tsv.
You can change the prefix of input files (tl_project.) using the argument -files_prefix <custom prefix>, you can also change the input/output path using -files_path <custom path>.
Output figures: tl_project.<orthogroup ID>.pdf.
Additional resources
For an example with data and practical usage examples, click here. For a detailed description of the theoretical background of this tool see references [1,2].
By following these steps and using REvolutionH-tl, you can conduct comprehensive evolutionary analysis tasks, including orthogroup selection, gene tree reconstruction, species tree reconstruction, and reconciliation, all in one powerful Python tool. Go to the section "Biological Relevance" for applications.
File format overview
For the example trees in this section, we'll use the example dataset (click here). In particular, we take the orthogroup inferred by REvolutionH-tl with the label 'OG7048'
Forbidden characters
Please avoid the usage of the following characters in the name of the species, genes, or other parameters of the tool: ;, /.
NHX format for trees
NHX format is a generalization of the Newick format. For example, the box below contains a species tree with 14 species as leaves. Note that every node of the three is associated with a node ID.
(((HUMAN[&&NHX:node_id=nID4],(MOUSE[&&NHX:node_id=nID6],RAT[&&NHX:node_id=nID7])[&&NHX:node_id=nID5])[&&NHX:node_id=nID3],(CHICK[&&NHX:node_id=nID9],DANRE[&&NHX:node_id=nID10])[&&NHX:node_id=nID8])[&&NHX:node_id=nID2],((ECOLI[&&NHX:node_id=nID13],((DICDI[&&NHX:node_id=nID16],((CAEEL[&&NHX:node_id=nID19],DROME[&&NHX:node_id=nID20])[&&NHX:node_id=nID18],ARATH[&&NHX:node_id=nID21])[&&NHX:node_id=nID17])[&&NHX:node_id=nID15],PLAF7[&&NHX:node_id=nID22])[&&NHX:node_id=nID14])[&&NHX:node_id=nID12],((SCHPO[&&NHX:node_id=nID25],YEAST[&&NHX:node_id=nID26])[&&NHX:node_id=nID24],CANAL[&&NHX:node_id=nID27])[&&NHX:node_id=nID23])[&&NHX:node_id=nID11])[&&NHX:node_id=nID1];
The box below contains a gene tree with 6 genes and two gene losses in the leaves. The label of loss leaves is 'X', and the leaves associated with existing genes have the attribute 'species'. Additionally, the label of inner nodes indicates evolutionary events; 'S' stands for 'speciation' and 'D' for 'duplication'. Finally, like in the species tree, every node of the three is associated with a node ID.
(((Phy001R8JZ[&&NHX:node_id=nID0:species=HUMAN],(Phy001RT94[&&NHX:node_id=nID5:species=MOUSE],Phy003F435[&&NHX:node_id=nID4:species=RAT])S[&&NHX:node_id=nID6])S[&&NHX:node_id=nID7],((Phy003I8P1[&&NHX:node_id=nID1:species=CHICK],Phy003ICTF[&&NHX:node_id=nID2:species=CHICK])D[&&NHX:node_id=nID3],X[&&NHX:node_id=nID9])S[&&NHX:node_id=nID8])S[&&NHX:node_id=nID10],((Phy003I6O9[&&NHX:node_id=nID11:species=CHICK],X[&&NHX:node_id=nID14])S[&&NHX:node_id=nID13],X[&&NHX:node_id=nID16])S[&&NHX:node_id=nID15])D[&&NHX:node_id=nID12];
You can use itol (Click here) for visualization of trees in nhx format. For example, the trees above look like this:

Reconciliation map format
An evolutionary scenario is a gene tree embedded inside a species tree. For this end, REvolutionH-tl constructs a reconciliation map going from nodes of the gene tree to nodes of the species tree.
The format for the reconciliation is the following: given the nodes x and y belonging to the gene and species trees correspondingly, we write the map from x to y as x:y. To include in a single string the map of all the nodes of a gene tree, we use a comma (,) as separator.
The tool uses the attribute node_id for node identification. The box below provides an example of reconciliation map between the gene and the species trees in the section above.
nID0:nID4,nID1:nID9,nID2:nID9,nID3:nID9,nID4:nID7,nID5:nID6,nID6:nID5,nID7:nID3,nID8:nID8,nID9:nID10,nID10:nID2,nID11:nID9,nID12:nID2,nID13:nID8,nID14:nID10,nID15:nID2,nID16:nID3
You can use the command revolutionhtl.plot_reconciliation for visualization of the reconciliation, as shown in the figure below. Note that markers at the inner nodes of the gene tree identify evolutionary events: blue diamond for duplication and red bullet for speciation. Similarly, markers at the leaves indicate if the gene exists in a current species (red bullet) or if the gene is extinct (black bullet).

Log file Every time you run REvolutionH-tl, the program writes in the file tl_project.log.txt the parameters used to run the program, as well as the time and progress.
Fasta directory Fasta files are required for steps 1 and 2. Specify this input using the attribute -F <directory>. You must have a fasta file for each species in your analysis (At least 2 species). The name of each file has to follow the format <species name>.fa. Please avoid the usage of forbidden characters in the name of the species.
By default, REvolutionH-tl searches for files with the extension ".fa", but you can change the extension using the attribute -fext <extension>.
Each gene in your analysis must have a unique identifier within and across species. Duplicated identifiers will raise an error. Remember to avoid forbidden characters in your identifiers.
Alignment hits directory This directory is the output of step 1, it has the suffix .alignment_all_vs_all/.
The alignment hits directory is required for step 2. Specify it using the attribute -alignment_h <directory>.
For each pair of species, you must have two files of alignment hits; from species one to species two, and from species two to species one. The name of each file has to follow the format <species_one>.vs.<species_two>.alignment_hits and has to be consistent with the name of fasta files.
REvolutionH-tl requires the alignment hits in tabular form with no headers. Each row of this table describes an alignment hit throughout seven columns. Below we describe those columns in the same way as Diamond and BLAST:
- qseqid: identifier of the query sequence.
- sseqid: identifier of the target sequence.
- qlen: length of the query sequence.
- slen: length of the target sequence.
- length: length of the alignment of the query sequence against the target sequence.
- bitscore: alignment statistic reflecting the degree of similarity of the target to the query sequence.
- evalue: alignment statistic reflecting significance. It is the number of subject sequences that can be expected to be retrieved from the database that have a bit score equal to or greater than the one calculated from the alignment of the query and subject sequence.
Orthogroups file This file is an output of step 2, it has the suffix .orthogroups.tsv.
Orthogroups are stored in a tabular format (.tsv file). Each row in this file represents one orthogroup. The first column assigns a unique identifier for each orthogroup. The number in the second column indicates the number of genes, and the third column shows the number of species represented in the orthogroup. The rest of the columns correspond to the species in your analysis, each of those columns contains the genes present in the orthogroup. If there is more than one gene per species, they are separated using a comma (',').
Best hits file This file is an output of step 2, it has the suffix .best_hits.tsv.
Best hits are required as input for step 3. You can provide them using the argument -best_h <.tsv file>.
Each row in this file describes a best hit. This file has to contain at least the following headers:
- OG: identifier of the orthogroup containing the genes of the hit.
- Query_species: species of the query gene.
- Target_species: species of the target gene
- Query_accession: identifier of the query gene.
- Target_accession: identifier of the target gene.
- Normalized_bit_score: normalized bit-score of the corresponding alignment hit.
Distances file This file is an output of step 2, it has the suffix .distances.tsv.
Distances are required as input for step 4. You can provide them using the argument -D <.tsv file>.
Each row in this table corresponds to a pair of genes. The header of the table must include:
- OG: identifier of the orthogroup containing the genes of the hit.
- Query_accession: identifier of the query gene.
- Target_accession: identifier of the target gene.
- score_distance: distance associated to the genes.
Gene trees files These files are generated at steps 3, 4, and 6. They have the suffixes .gene_trees.tsv, .resolved_trees.tsv, .corrected_trees.tsv, and .reconcilied_trees.tsv.
Gene trees are input of steps 4, 5, and 6. You can provide them using the argument -T <.tsv file>. Note that gene trees are inferred at step 3 and further processed at steps 4 (tree refinement) and 6 (tree reconciliation), we recall whenever REvolutionH-tl has to decide between manually specified files and files produced at a previous step, the tool prioritizes the second option.
Each row in these tables contains the information for one gene tree. It has to contain the columns:
- OG: identifier of the orthogroup containing the genes of the hit.
- tree: tree in nhx format
The leaves of a gene tree are gene identifiers or the loss label 'X'. Leaves also have the attribute "species". The species of the genes must be consistent with the species tree. The inner nodes of the gene tree are associated with evolutionary events: the letter "S" indicates a speciation event, while "D" stands for gene duplication. Additionally, for reconciliation, gene and species trees have to include the attribute 'node_ID' for all the nodes.
Furthermore, each file contains additional columns showing information added at each of the steps:
-
.gene_trees.tsv
Step 3 of REvolutionH-tl constructs gene trees by analysis of best-hit relationships. This file only contains the two columns mentioned above.
-
.resolved_trees.tsv
Step 4 resolves duplication nodes. This file includes the columns:
- original_polytomies: number of polytomies present at the input trees.
- resolved_polytomies: number of successfully resolved polytomies.
-
.corrected_trees.tsv
Step 6 checks for consistency between gene and species tree before reconciliation. Inconsistent trees are edited by pruning conflicting genes. This file includes the columns:
- inconsistent_triples: number of inconsistent triples present in the input trees.
- prunned_leaves: list of genes pruned from the input trees.
-
.reconcilied_trees.tsv
Step 6 reconciles gene and species trees by construction of a reconciliation map. This file contains the column:
- reconciliation_map: map going from nodes of the gene tree to nodes of the species tree as specified in section 'Reconciliation map format'.
Orthologs file This file is an output of step 3, it has the suffix .orthologs.tsv.
Orthology is stored in a tabular form. Each row of this table contains an orthology relation, i.e. the information of a pair of orthologous genes, described in 6 columns:
- a, b: pair of orthologous genes
- species_a, species_b: species corresponding to the orthologous genes.
- OG: identifier of the orthogroup containing the orthologous genes.
- Normalized_bit_score: normalized bit-score associated with the best hits between genes a and b. If there is not a best hit, we place the symbol "*".
Best matches file This file is an output of step 3, it has the suffix .best_matches.tsv.
Best matches are stored in a tabular form. Each row of this table contains the information of a best-match throughout 5 columns:
- OG: identifier of the orthogroup containing the best match.
- Query_accession: query gene.
- Query_species: species of the query gene.
- Target_accession: a best match of the query gene in the target species.
- Target_species: species of the target gene.
Species tree file This file is an output of steps 5 and 6, they have the suffixes .species_tree.tsv and .labeled_species_tree.nhx correspondingly.
The species tree is required for step 6. You can provide it using the argument -S <.nhxx file>.
The species tree has species as leaves. The name of the species must be consistent with the species of the genes in the gene trees. Additionally, the tree output at step 6 has the attribute 'node_id' for every node, specifying a unique identifier for each node of the tree.
Biological Relevance
RevolutionH-tl is a versatile tool that facilitates various aspects of evolutionary and comparative genomics research. Its outputs are useful for advancing our understanding of the evolution and functional roles of genes across different species. It has several biological applications:
- Phylogenetics: RevolutionH-tl aids in constructing accurate gene and species trees, allowing researchers to infer the evolutionary history of genes and species. This information is fundamental for understanding the relationships among different organisms.
- Functional Genomics: By identifying orthogroups and best hits, RevolutionH-tl helps researchers discover homologous genes with similar functions. This can be invaluable for functional annotation and comparative genomics studies.
- Evolutionary Genomics: Researchers can use RevolutionH-tl to explore gene duplication and speciation events. This information sheds light on the evolutionary processes that have shaped gene families and species.
- Biological Databases: The output files from RevolutionH-tl can be integrated into biological databases to enhance the annotation of genes and improve our understanding of gene and species relationships.
- Phylogenomic Analyses: RevolutionH-tl's ability to reconcile gene trees with species trees provides insights into the complex interplay between gene duplication, loss, and speciation. It is invaluable for conducting phylogenomic analyses.
- Comparative Genomics: Researchers can compare the output from RevolutionH-tl across different species to identify conserved genes and understand how specific genes have evolved in different lineages.
- Functional Inference: The orthologous relationships identified by REvolutionH-tl can be used to infer gene function by transferring functional annotations from well-characterized genes to orthologs.
References
[1] Ramírez-Rafael J. A., Korchmaros A., Aviña-Padilla K., López-Sánchez A., Martinez-Medina G., Hernández-Álvarez A., Hellmuth M., Stadler P. F., and Hernandez-Rosales M. (2025) REvolutionH-tl: A Fast and Robust Tool for Decoding Evolutionary Gene Histories. Submitted.
[2] Ramírez-Rafael J. A., Korchmaros A., Aviña-Padilla K., López-Sánchez A., España-Tinajero A. A., Hellmuth M., Stadler P. F., and Hernandez-Rosales M. (2024) REvolutionH-tl: Reconstruction of Evolutionary Histories tool. In Comparative Genomics: 21st International Conference, RECOMB-CG 2024, Boston, MA, USA, April 27–28, 2024, Proceedings. Springer-Verlag, Berlin, Heidelberg, 89–109. https://doi.org/10.1007/978-3-031-58072-7_5
[3] Buchfink B, Reuter K, Drost HG, "Sensitive protein alignments at tree-of-life scale using DIAMOND", Nature Methods 18, 366–368 (2021). doi:10.1038/s41592-021-01101-x
[4] Fassler J, Cooper P. BLAST Glossary. 2011 Jul 14. In: BLAST® Help [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2008-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK62051/