
Security News
The Hidden Blast Radius of the Axios Compromise
The Axios compromise shows how time-dependent dependency resolution makes exposure harder to detect and contain.
By Ahmad Nassri - Apr 01, 2026
savemoney
Advanced tools
Simple Algorithm for Very Efficient Multiplexing of Oxford Nanopore Experiments for You!
![]()
Simple Algorithm for Very Efficient Multiplexing of Oxford Nanopore Experiments for You!
SAVEMONEY guides researchers to mix multiple plasmids for submission as a single sample to a commercial long-read sequencing service (e.g., Oxford Nanopore Technology), reducing overall sequencing costs while maintaining fidelity of sequencing results. Following is the outline of the procedure:

The algorithm permits mixing of six (or potentially even more) plasmids for sequencing with Oxford Nanopore Technology (e.g., Plasmidsaurus services) and permits mixing of plasmids with as few as two base differences. For more information, please check out our publication (coming soon).
Verified on macOS, Linux, and Windows10
SAVEMONEY is available via pip.
pip install savemoney
If installation via pip fails, please check the requirements above. If any of the package conflicts with those already present in your environment, I recommend creating a new virtual environment.
If C++ compiler does not exist, install Xcode Command Line Tools using the following command (for macOS):
xcode-select --install
or download Microsoft C++ Build Tools (for Windows).
SAVEMONEY can be executed either in the python script or via command line.
To import and execute SAVEMONEY in the python script. Follow the example below:
import savemoney
savemoney.pre_survey("path_to_sequence_directory", "save_directory", **kwargs)
savemoney.post_analysis("path_to_sequence_directory", "save_directory", **kwargs)
All of the plasmid map files with *.dna and .fasta extension (and in addition *.fastq files for post analysis) in the path_to_sequence_directory will be used for the analysis. Results will be generated in the save_directory. kwargs are optional parameters through which you can optimize the analysis:
# pre-survey
kwargs = {
'distance_threshold': 5, # main parameter to be changed
'number_of_groups': 1, # main parameter to be changed
'gap_open_penalty': 3, # alignment parameter
'gap_extend_penalty': 1, # alignment parameter
'match_score': 1, # alignment parameter
'mismatch_score': -2, # alignment parameter
'topology_of_dna': 0, # 0: circular, 1: linear
'n_cpu': 2, # number of cpu cores to be used
'export_image_results': 1, # 0; skip export of svg figure files, 1: export svg figure files
}
# post-analysis
kwargs = {
'score_threshold': 0.3, # main parameter to be changed
'gap_open_penalty': 3, # alignment parameter
'gap_extend_penalty': 1, # alignment parameter
'match_score': 1, # alignment parameter
'mismatch_score': -2, # alignment parameter
'error_rate': 0.00001, # prior probability for Bayesian analysis
'ins_rate': 0.00001, # prior probability for Bayesian analysis
'window': 160, # maximum detectable length of repetitive sequences when wrong plasmid maps are provided: if region of 80 nt is repeated adjascently two times, put the value of 160
'topology_of_dna': 0, # 0: circular, 1: linear
'n_cpu': 2, # number of cpu cores to be used
'export_image_results': 1, # 0; skip export of svg figure files, 1: export svg figure files
}
For the meaning of these parameters, please refer to the SAVEMONEY Google Colab page or the reference below.
SAVEMONEY can also be executed via command line:
python -m savemoney.pre_survey path_to_sequence_directory save_directory
python -m savemoney.post_analysis path_to_sequence_directory save_directory
Parameters can be specified as follows:
# pre-survey
python -m savemoney.pre_survey -h
usage: __main__.py [-h] [-gop GOP] [-gep GEP] [-ms MS] [-mms MMS] [-dt DT] [-nog NOG] plasmid_map_dir_paths save_dir_base
positional arguments:
plasmid_map_dir_paths path to plasmid map_directory
save_dir_base save directory path
options:
-h, --help show this help message and exit
-gop GOP gap_open_penalty, optional, default_value = 3
-gep GEP gap_extend_penalty, optional, default_value = 1
-ms MS match_score, optional, default_value = 1
-mms MMS mismatch_score, optional, default_value = -2
-dt DT distance_threshold, optional, default_value = 5
-nog NOG number_of_groups, optional, default_value = 1
-tod TOD topology_of_dna, optional, default_value = 0 (0: circular, 1: linear)
-nc NC n_cpu, optional, default_value = 2
-eir EIR export_image_results, optional, default_value = 1
# post-analysis
python -m savemoney.post_analysis -h
usage: __main__.py [-h] [-gop GOP] [-gep GEP] [-ms MS] [-mms MMS] [-st ST] [-er ER] [-dmr DMR] [-ir IR] sequence_dir_paths save_dir_base
positional arguments:
sequence_dir_paths sequence_dir_paths
save_dir_base save directory path
options:
-h, --help show this help message and exit
-gop GOP gap_open_penalty, optional, default_value = 3
-gep GEP gap_extend_penalty, optional, default_value = 1
-ms MS match_score, optional, default_value = 1
-mms MMS mismatch_score, optional, default_value = -2
-st ST score_threshold, optional, default_value = 0.3
-er ER error_rate, optional, default_value = 1e-07
-ir IR ins_rate, optional, default_value = 1e-07
-w W window, optional, default_value = 160
-tod TOD topology_of_dna, optional, default_value = 0 (0: circular, 1: linear)
-nc NC n_cpu, optional, default_value = 2
-eir EIR export_image_results, optional, default_value = 1
The interpretation of output files are described on SAVEMONEY Google Colab page in details. Other than that, you can visualize consensus alignment results by using your_plasmid_name.ca file generated by SAVEMONEY.
From python script:
import savempney
savemoney.show_consensus(consensus_alignment_path, center=2000, seq_range=50, offset=0)
From command line:
python -m savemoney.show_consensus path_to_consensus_alignment_file
Parameters can be specified as follows:
python -m savemoney.show_consensus -h
usage: __main__.py [-h] [--center CENTER] [--seq_range SEQ_RANGE] [--offset OFFSET] consensus_alignment_path
positional arguments:
consensus_alignment_path path to consensus_alignment (*.ca) file
options:
-h, --help show this help message and exit
--center CENTER center, optional, default_value = 2000
--seq_range SEQ_RANGE seq_range, optional, default_value = 50
--offset OFFSET offset, optional, default_value = 0
Conversion of consensus alignment results (*.ca) to *.bam and *.fastq format is also supported. The conversion requires pysam>=0.22.0 be installed in your environment. To convert the file, type the following code in a python script:
import savemoney
savemoney.ca2bam(consensus_alignment_path)
If you want to convert it via command line, type the following commnad:
python -m savemoney.ca2bam path_to_consensus_alignment_file
FAQs
Simple Algorithm for Very Efficient Multiplexing of Oxford Nanopore Experiments for You!
We found that savemoney demonstrated a healthy version release cadence and project activity because the last version was released less than a year ago. It has 1 open source maintainer collaborating on the project.
Did you know?

Socket for GitHub automatically highlights issues in each pull request and monitors the health of all your open source dependencies. Discover the contents of your packages and block harmful activity before you install or update your dependencies.
Security News
The Axios compromise shows how time-dependent dependency resolution makes exposure harder to detect and contain.
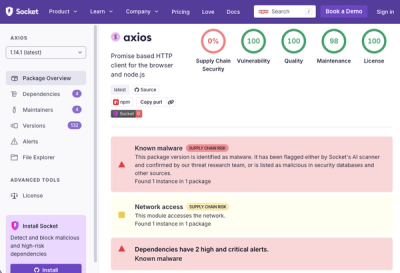
Research
A supply chain attack on Axios introduced a malicious dependency, plain-crypto-js@4.2.1, published minutes earlier and absent from the project’s GitHub releases.

Research
Malicious versions of the Telnyx Python SDK on PyPI delivered credential-stealing malware via a multi-stage supply chain attack.