
Security News
The Hidden Blast Radius of the Axios Compromise
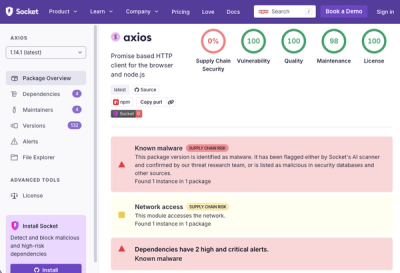
The Axios compromise shows how time-dependent dependency resolution makes exposure harder to detect and contain.
By Ahmad Nassri - Apr 01, 2026
snapper-av
Advanced tools
![]()

![]()
SNAPPER is a method designed to adjust introns in spliced alignments to best match their reference transcriptomic positions. Aligners such as minimap2 are built to only consider guide annotations on the genome to which sequences are being aligned to. However, in case of transcript alignments where sequences have often been extracted from the reference genome using an annotation, there is an additional unused prior in the form of reference exon boundaries in the sequences themselves. it is only logical to use this prior information to guide the alignment, alas such functionality is not present in most aligners. To circumvent this issue, SNAPPER was developed to adjust the intron positions in the alignment to best match the reference transcriptomic positions. The method reconstructs the alignment DP matrix, augments it with weights for reference intron positions and performs a traceback penalizing mismatches with respect to the original alignment and prioritizing intron positions from the reference annotation. The result is a more consistent alignment especially when it comes to alignment between poorly conserved genomes.
Coming soon...
The easiest way to install SNAPPER is through PyPI:
$ pip install snapper-av
$ snapper --help
To uninstall SNAPPER:
$ pip uninstall snapper-av
To build from source, clone the git repository:
$ git clone https://github.com/alevar/snapper.git --recursive
$ cd snapper
$ pip install -r requirements.txt
$ pip install .
| Requirement | Details |
|---|---|
| Language support | Python ≥ 3.6 |
| Dependencies | - |
snapper [-h] -s SAM -r REFERENCE [-o OUTPUT] [--qry_intron_match_score QRY_INTRON_MATCH_SCORE]
[--trg_pos_match_score TRG_POS_MATCH_SCORE] [--trg_pos_mismatch_score TRG_POS_MISMATCH_SCORE]
| Option | Description |
|---|---|
-s, --sam | Path to the SAM/BAM alignment file. Read names in the alignment are expected to match corresponding transcript_id in the reference annotationPath to the query GTF/GFF annotation file. |
-r, --reference | Path to the reference annotation. transcript_id field is expected to match read names in the sam alignment. |
-o, --output | Path to the output SAM/BAM file. |
--qry_intron_match_score | Score for matching query introns. |
--trg_pos_match_score | Score for matching target positions. |
--trg_pos_mismatch_score | Score for mismatching target positions. |
| Option | Description |
|---|---|
-h, --help | Prints help message. |
Sample datasets are provided in the "example" directory to test and get familiar with SNAPPER.
The included example can be run with the following command from the root directory of the repository:
snapper --sam example/example.gtf --reference example/example.gtf --output example/output.sam
FAQs
SNAPPER: Correct Intron Shifts in Alignments via Reference Annotation
We found that snapper-av demonstrated a healthy version release cadence and project activity because the last version was released less than a year ago. It has 1 open source maintainer collaborating on the project.
Did you know?

Socket for GitHub automatically highlights issues in each pull request and monitors the health of all your open source dependencies. Discover the contents of your packages and block harmful activity before you install or update your dependencies.
Security News
The Axios compromise shows how time-dependent dependency resolution makes exposure harder to detect and contain.
Research
A supply chain attack on Axios introduced a malicious dependency, plain-crypto-js@4.2.1, published minutes earlier and absent from the project’s GitHub releases.

Research
Malicious versions of the Telnyx Python SDK on PyPI delivered credential-stealing malware via a multi-stage supply chain attack.