Product
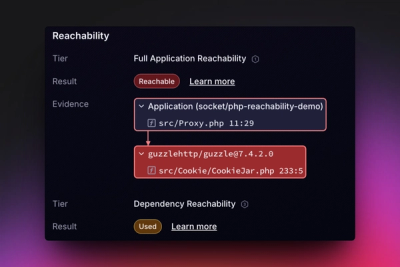
Introducing Reachability for PHP
Reachability analysis for PHP is now available in experimental, helping teams identify which vulnerabilities are actually exploitable.
By Benjamin Barslev - Apr 24, 2026
bio-shark
Advanced tools





To accurately assess homology between unalignable sequences, we developed an alignment-free sequence comparison algorithm, SHARK (Similarity/Homology Assessment by Relating K-mers).
We trained SHARK-dive, a machine learning homology classifier, which achieved superior performance to standard alignment in assessing homology in unalignable sequences, and correctly identified dissimilar IDRs capable of functional rescue in IDR-replacement experiments reported in the literature.
Scoring the similarity between a pair of sequence
Variants:
SHARK-score (T))SHARK-score (best))Find sequences similar to a given query from a target set
Find conserved motifs (k-mers) amongst a set of (similar) sequences
SHARK officially supports Python versions >=3.8,<3.13.
Recommended Use within a local Python virtual environment
python3 -m venv /path/to/new/virtual/environment
The collection of SHARK tools is available in PyPI and can be installed via pip. Versions <2.0.0 include SHARK-Dive and SHARK-Score only. From version >=2.0.0 on, SHARK-capture is also included.
$ pip install bio-shark
$ git clone git@git.mpi-cbg.de:tothpetroczylab/shark.git
Once you have a copy of the source, you can embed it in your own Python package, or install it into your site-packages easily.
# Make sure you have the required Python version installed
$ python3 --version
Python 3.11.5
$ cd shark
$ python3 -m venv shark-env
$ source shark-env/bin/activate
$ (shark-env) % python -m pip install .
$ pip install git+https://git.mpi-cbg.de/tothpetroczylab/shark.git
shark-score along with input fasta files and scoring parameters% shark-score QUERYSEQUENCE TARGETSEQUENCE -k 5 - t 0.95 -s threshold -o results.tsv
% shark-score --infile <path/to/query/fasta/file> --dbfile <path/to/target/fasta/file> --outfile <path/to/result/file> --length <k-mer length> --threshold <shark-score threshold>
usage: shark-score [-h] [--infile INFILE] [--dbfile DBFILE] [--outfile OUTFILE] [--scoretype {best,threshold,NGD}] [--length LENGTH] [--threshold THRESHOLD] [query] [target]
Run SHARK-Scores (best or T=x variants) or Normalised Google Distance Scores. Note that if a FASTA file is provided, it will be used instead.
positional arguments:
query Query sequence
target Target sequence
optional arguments:
-h, --help show this help message and exit
--infile INFILE, -i INFILE
Query FASTA file
--dbfile DBFILE, -d DBFILE
Target FASTA file
--outfile OUTFILE, -o OUTFILE
Result file
--scoretype {best,threshold,NGD}, -s {best,threshold,NGD}
Score type: best or threshold or NGD. Default is threshold.
--length LENGTH, -k LENGTH
k-mer length
--threshold THRESHOLD, -t THRESHOLD
threshold for SHARK-Score (T=x) variant
from bio_shark.dive import run
result = run.run_normal(
sequence1="LASIDPTFKAN",
sequence2="ERQKNGGKSDSDDDEPAAKKKVEYPIAAAPPMMMP",
k=3,
threshold=0.8
)
print(result)
0.2517953859
# `run_sparse` and `run_collapsed` have similar input structure except for `threshold`
from bio_shark.dive.prediction import Prediction
from bio_shark.core import utils
id_sequence_map1 = utils.read_fasta_file(file_path="<absolute-file-path-query-fasta>")
id_sequence_map2 = utils.read_fasta_file(file_path="<absolute-file-path-target-fasta>")
predictor = Prediction(q_sequence_id_map=id_sequence_map1, t_sequence_id_map=id_sequence_map2)
output = predictor.predict() # List of output objects; Each element is for one pair
shark-dive with the absolute path of the sequence fasta files as only argumentprediction is always based on scores of k = [1..10]sample_fasta_file.fasta from data folder (Owncloud link)usage: shark-dive [-h] [--output_dir OUTPUT_DIR] query target
DIVE-Predict: Given some query sequences, compute their similarity from the list of target sequences;Target is
supposed to be major database of protein sequences
positional arguments:
query Absolute path to fasta file for the query set of input sequences
target Absolute path to fasta file for the target set of input sequences
options:
-h, --help show this help message and exit
--output_dir OUTPUT_DIR
Output folder (default: current working directory)
$ shark-dive "<query-fasta-file>.fasta" "<target-fasta-file>.fasta"
Read fasta file from path <query-fasta-file>.fasta; Found 4 sequences; Skipped 0 sequences for having X
Read fasta file from path <target-fasta-file>.fasta; Found 6 sequences; Skipped 0 sequences for having X
Output stored at <OUTPUT_DIR>/<path-to-sequence-fasta-file>.fasta.csv
$ bash parallel_run.sh
...
Read fasta file from path ../data/IDR_Segments.fasta; Found 6 sequences; Skipped 0 sequences for having non-canonical AAs
All sequences are present! Proceeding with SHARK-dive prediction...
Finished in 0.10163092613220215 seconds
121307136
SHARK-dive prediction complete!
Elapsed Time: 3 seconds
Refer to this READme as an example to run capture using multi-processing using capture/compute.py
Refer to capture/compute_slurm/README.md to run CAPTURE pipeline on an HPC (using the SLURM workload manager)
Run the Python command shark-capture with the following positional arguments:
Optional arguments include
Note that, in the command-line run, folders are automatically created and named according to sharkcapture default naming conventions. Hadamard matrices are stored in the hadamard_{k-mer_length} folder as all_hadamards.json_all and conserved k-mers are stored in the conserved_kmers folder as k_{k-mer length}.json
Example:
$ shark-capture -h
usage: shark-capture [-h] [--outfile OUTFILE] [--k_min K_MIN] [--k_max K_MAX] [--n_output N_OUTPUT]
[--n_processes N_PROCESSES] [--log] [--extend] [--cutoff CUTOFF]
[--no_per_sequence_kmer_plots | --sequence_subset SEQUENCE_SUBSET]
sequence_fasta_file_path output_dir
SHARK-capture: An alignment-free, k-mer x similarity-based motif detection tool
positional arguments:
sequence_fasta_file_path
Absolute path to fasta file of input sequences
output_dir Output folder path
options:
-h, --help show this help message and exit
--outfile OUTFILE name of consensus k-mers output file
--k_min K_MIN Min k-mer length of captured motifs
--k_max K_MAX Max k-mer length of captured motifs
--n_output N_OUTPUT number of top consensus k-mers to output and process for subsequent steps
--n_processes N_PROCESSES
No. of processes (python multiprocessing
--log flag to show scores in log scale (base 10) for per-sequence k-mer matches plot
--extend enable SHARK-capture Extension Protocol
--cutoff CUTOFF Percentage cutoff for SHARK-capture Extension Protocol, default 0.9
--no_per_sequence_kmer_plots
flag to suppress plotting of per-sequence k-mer matches.Mutually exclusive with
--sequence_subset
--sequence_subset SEQUENCE_SUBSET
comma separated sequence identifiers or substrings to generate output for per-sequence
k-mer matches plot, e.g. "sequence_id_1,sequence_id_2". By default, plots for all
sequences. Mutually exclusive with --no_per_sequence_kmer_plots.
$ shark-capture "<query-fasta-file>.fasta" "<output-directory-name>" --n_output 20 --outfile top20.txt
Read fasta file from path <query-fasta-file>.fasta; Found 4 sequences; Skipped 0 sequences for having X
Processing K=3
Collected args (sequence pairs): 36046
k=3 - Created master input data file at <output-directory-name>/input_params/k_3.json
Completed processing. Gathered hadamard reciprocals for: 36046
Hadamard sorted k-mer score mapping stored at <output-directory-name>/hadamard_3/all_hadamards.json_all
Search space (no. unique k-mers): 2684
Hadamard sorted k-mer score mapping stored at <output-directory-name>/conserved_kmers/k_3.json
...
Processing K=10
Collected args (sequence pairs): 36046
k=10 - Created master input data file at <output-directory-name>/input_params/k_10.json
Completed processing. Gathered hadamard reciprocals for: 36046
Hadamard sorted k-mer score mapping stored at <output-directory-name>/hadamard_10/all_hadamards.json_all
Search space (no. unique k-mers): 15069
Hadamard sorted k-mer score mapping stored at <output-directory-name>/conserved_kmers/k_10.json
Reporting top 20 K-Mers, stored in top20.txt
SHARK-capture completed successfully! All outputs stored in <output-directory-name>
The main outputs are:
a comma-separated, ranked table of the top consensus k-mers and their corresponding shark-capture score as sharkcapture_consensus_kmers.txt
a tab-separated table for each consensus k-mer listing the occurrences of the best reciprocal match (if found) in each sequence as sharkcapture_{consensus_k-mer}_occurrences.tsv. The columns indicate (in order):
a probability matrix generated from the matched occurrence table for each consensus k-mer as {consensus_k-mer}_probabilitymatrix.csv
a sequence-logo-like visual representation of the conservation (information content) of each consensus k-mer, generated from the probability matrix as {consensus_k-mer}_logo.png
$ docker build . -f Dockerfile -t atplab/bio-shark
# Run shark-capture with the help option to start with
$ docker run atplab/bio-shark -h
# You also need to map inputs and outputs volumes from inside the container to your local file system, when you run the docker service
$ docker run -v <absolute-path-to-file-on-local-machine>/IDR_Segments.fasta:/app/inputs/IDR_Segments.fasta \
-v <absolute-path-to-file-on-local-machine>/outputs:/app/outputs \
atplab/bio-shark /app/inputs/IDR_Segments.fasta outputs
$ docker run -it --entrypoint sh atplab/bio-shark
Examples of how to use and run SHARK are shown in a provided Jupyter notebook. The notebook can be found under the notebooks folder.
Please read documentation here.
Create a virtual environment by executing the command venv:
$ python -m venv /path/to/new/virtual/environment
# e.g.
$ python -m venv my_jupyter_env
Then install the classic Jupyter Notebook and the seaborn dependency with:
$ source my_jupyter_env/bin/activate
$ pip install notebook seaborn
Also install bio-shark from source in the same virtual environment...
$ pip install .
Finally create a new Kernel using ipykernel...
python -m ipykernel install --user --name my_jupyter_env --display-name "Python (my_jupyter_env)"
In your terminal source the previously created virtual environment...
$ source my_jupyter_env/bin/activate
Launch Jupyter Notebook...
$ jupyter notebook
In the jupyter browser GUI, open the example notebook called 'dive_feature_viz.ipynb' under the notebooks folder.
Once that is done, change the kernel in the GUI, before your execute the notebook itself. This will make sure you operate on the correct virtual Python environment, which contains all required dependencies like for instance, seaborn.

SHARK enables sensitive detection of evolutionary homologs and functional analogs in unalignable and disordered sequences. Chi Fung Willis Chow, Soumyadeep Ghosh, Anna Hadarovich, and Agnes Toth-Petroczy. Proc Natl Acad Sci U S A. 2024 Oct 15;121(42):e2401622121. doi: 10.1073/pnas.2401622121. Epub 2024 Oct 9. PMID: 39383002.

This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
FAQs
SHARK (Similarity/Homology Assessment by Relating K-mers)
We found that bio-shark demonstrated a healthy version release cadence and project activity because the last version was released less than a year ago. It has 0 open source maintainers collaborating on the project.
Did you know?

Socket for GitHub automatically highlights issues in each pull request and monitors the health of all your open source dependencies. Discover the contents of your packages and block harmful activity before you install or update your dependencies.
Product
Reachability analysis for PHP is now available in experimental, helping teams identify which vulnerabilities are actually exploitable.
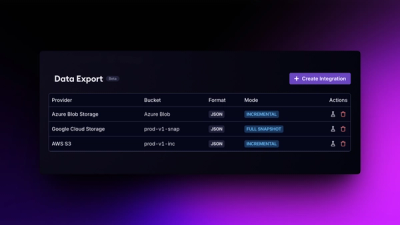
Product
Export Socket alert data to your own cloud storage in JSON, CSV, or Parquet, with flexible snapshot or incremental delivery.
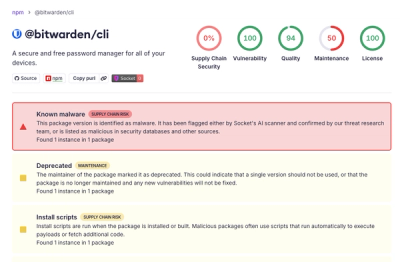
Research
/Security News
Bitwarden CLI 2026.4.0 was compromised in the Checkmarx supply chain campaign after attackers abused a GitHub Action in Bitwarden’s CI/CD pipeline.