
Company News
Socket Has Acquired Secure Annex
Socket has acquired Secure Annex to expand extension security across browsers, IDEs, and AI tools.
By Feross Aboukhadijeh - Apr 28, 2026
pyjess
Advanced tools

Cython bindings and Python interface to Jess, a 3D template matching software.


![]()






![]()
![]()


![]()
Jess is an algorithm for constraint-based structural template matching proposed by Jonathan Barker et al.[1]. It can be used to identify catalytic residues from a known template inside a protein structure. Jess is an evolution of TESS, a geometric hashing algorithm developed by Andrew Wallace et al.[2], removing some pre-computation and structural requirements from the original algorithm. Jess was further updated and maintained by Ioannis Riziotis during his PhD in the Thornton group.
PyJess is a Python module that provides bindings to Jess using Cython. It allows creating templates, querying them with protein structures, and retrieving the hits using a Python API without performing any external I/O. It's also more than 10x faster than Jess thanks to algorithmic optimizations added to improve the original Jess code while producing consistent results. PyJess was developed to support EnzyMM, the Enzyme Motif Miner[3].
PyJess is available for all modern Python versions (3.7+).
It can be installed directly from PyPI, which hosts some pre-built x86-64 wheels for Linux, MacOS, and Windows, as well as the code required to compile from source with Cython:
$ pip install pyjess
Otherwise, PyJess is also available as a Bioconda package:
$ conda install -c bioconda pyjess
Check the install page of the documentation for other ways to install PyJess on your machine.
PyJess is scientific software, published part of the EnzyMM[3] publication, and builds on top of Jess[2]. Please cite also Jess if you are using PyJess in an academic work, for instance as:
PyJess (Hackett et al., 2026), a Python library binding to Jess (Barker et al., 2003).
Load Template
objects to be used as references from different template files:
import pathlib
import pyjess
templates = []
for path in sorted(pathlib.Path("vendor/jess/examples").glob("template_*.qry")):
templates.append(pyjess.Template.load(path, id=path.stem))
Load a Molecule
(a PDB structure) from a PDB file, create one with the Python API, or
convert it from a Bio.Model,
gemmi.Model,
or biotite.structure.AtomArray
object:
# load from PDB file or mmCIF file
mol = pyjess.Molecule.load("vendor/jess/examples/test_pdbs/pdb1a0p.ent")
# load with BioPython
parser = Bio.PDB.PDBParser()
structure = parser.get_structure('pdb1a0p', "vendor/jess/examples/test_pdbs/pdb1a0p.ent")
mol = Molecule.from_biopython(structure, id="1a0p")
# load with Gemmi
structure = gemmi.read_pdb_string("vendor/jess/examples/test_pdbs/pdb1a0p.ent")
mol = Molecule.from_gemmi(structure[0], id="1a0p")
# load with Biotite
pdb_file = biotite.structure.io.pdb.PDBFile.read(f)
structure = pdb_file.get_structure(altloc="all", extra_fields=["atom_id", "b_factor", "occupancy", "charge"])
mol = Molecule.from_biotite(structure[0])
Create a Jess
instance and use it to query a against the stored templates:
jess = pyjess.Jess(templates)
query = jess.query(mol, rmsd_threshold=2.0, distance_cutoff=3.0, max_dynamic_distance=3.0)
The hits are computed iteratively, and the different output statistics are computed on-the-fly when requested:
for hit in query:
print(hit.molecule().id, hit.template().id, hit.rmsd, hit.log_evalue)
for atom in hit.atoms():
print(atom.name, atom.x, atom.y, atom.z)
Hits can also be rendered in PDB format like in the original Jess output, either by writing to a file directly, or to a Python string:
for hit in query:
hit.dump(sys.stdout, format="pdb")
Once a Jess
instance has been created, the templates cannot be edited anymore,
making the Jess.query method re-entrant and thread-safe. This allows querying
several molecules against the same templates in parallel using e.g a
ThreadPool:
molecules = []
for path in glob.glob("vendor/jess/examples/test_pdbs/*.ent"):
molecules.append(Molecule.load(path))
with multiprocessing.ThreadPool() as pool:
hits = pool.map(jess.query, molecules)
⚠️ Prior to PyJess v0.2.1, the Jess code was running some thread-unsafe operations which have now been patched.
If running Jess in parallel, make sure to use v0.2.1 or later to use the code patched with re-entrant functions.
The following table reports the runtime of PyJess to match N=132 protein structures to the M=7607 templates of EnzyMM, using J=12 threads to parallelize.
| Version | Runtime (s) | Match Speed (N * M / s * J) | Speedup |
|---|---|---|---|
v0.4.2 | 618.1 | 135.4 | N/A |
v0.5.0 | 586.3 | 142.7 | x1.05 |
v0.5.1 | 365.6 | 228.9 | x1.69 |
v0.5.2 | 327.2 | 255.7 | x1.88 |
v0.6.0 | 54.5 | 1535.4 | x11.34 |
v0.7.0 | 52.4 | 1597.5 | x11.80 |
Benchmarks were run on a quiet i7-1255U CPU running @4.70GHz with 10 physical cores / 12 logical cores.
Found a bug ? Have an enhancement request ? Head over to the GitHub issue tracker if you need to report or ask something. If you are filing in on a bug, please include as much information as you can about the issue, and try to recreate the same bug in a simple, easily reproducible situation.
Contributions are more than welcome! See
CONTRIBUTING.md
for more details.
This project adheres to Semantic Versioning and provides a changelog in the Keep a Changelog format.
This library is provided under the MIT License. The JESS code is distributed under the MIT License as well.
This project is in no way not affiliated, sponsored, or otherwise endorsed by the JESS authors. It was developed by Martin Larralde during his PhD project at the Leiden University Medical Center in the Zeller team.
FAQs
Cython bindings and Python interface to JESS, a 3D template matching software.
We found that pyjess demonstrated a healthy version release cadence and project activity because the last version was released less than a year ago. It has 2 open source maintainers collaborating on the project.
Did you know?

Socket for GitHub automatically highlights issues in each pull request and monitors the health of all your open source dependencies. Discover the contents of your packages and block harmful activity before you install or update your dependencies.
Company News
Socket has acquired Secure Annex to expand extension security across browsers, IDEs, and AI tools.

Research
/Security News
Socket is tracking cloned Open VSX extensions tied to GlassWorm, with several updated from benign-looking sleepers into malware delivery vehicles.
Product
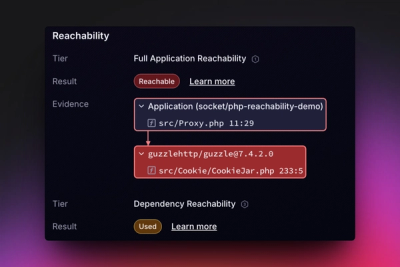
Reachability analysis for PHP is now available in experimental, helping teams identify which vulnerabilities are actually exploitable.