Product
Announcing Precomputed Reachability Analysis in Socket
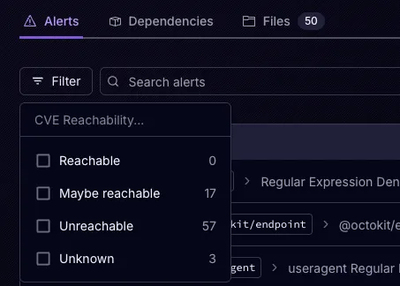
Socket’s precomputed reachability slashes false positives by flagging up to 80% of vulnerabilities as irrelevant, with no setup and instant results.
By Martin Torp - Jul 30, 2025
A standalone bacterial amplicon designing tool (Tuberculosis Optimized Amplicon Sequencing Tool)
We present TOAST, a software tool designed to streamline and optimize amplicon primer design for Mycobacterium tuberculosis sequencing. TOAST integrates the robust primer design capabilities of Primer3—accounting for Tm, homopolymers, hairpins, and homodimers—with an in-house pipeline that rigorously filters for heterodimer formation and unintended alternative binding. What sets TOAST apart is its automation and intelligence: it leverages a curated database of over 50 M. tuberculosis genomes to inform amplicon placement, ensuring robust primer performance across strain diversity. Users can prioritize SNPs for coverage, focus on specific resistance genes, and tailor designs to spoligotype backgrounds. The tool outputs primer sequences along with detailed thermodynamic profiles and genomic coordinates, making it an end-to-end solution for targeted TB panel design.
toast design)Main Inputs:
-s): Default is globally collated clinical TB SNPs-ref): Default is MTB-h37rv genome-sp_f)-ud) and custom genomic features (-gff)Configurable Settings:
-a)-p): Default is amplicon size divided by 6-sn) and targeted gene names (-sg)-nn)-g) to visualize amplicon coverage-all_snp) uses all frequent SNPs from the default database to design degenerate primers. For other species, a custom SNP file in the same format can be provided to override the default.
Outputs:-op)Example Usage:
toast design -op ./output -a 400 -sn 2 -sg rpoB,katG -nn 20
This command designs amplicons of 400 base pairs, including two specifically targeting the rpoB and katG genes, and 20 additional amplicons for prioritized SNP coverage.
toast amplicon_no)Purpose:
Estimate the number of amplicons required to achieve desired SNP coverage in TB genomic studies.
Main Inputs:
-s)-ref)Settings:
-a)-g)Outputs:
Estimates and coverage graphics saved in the specified output directory (-op).
toast plotting)Purpose:
Visualize and analyze the coverage and distribution of designed amplicons.
Main Inputs:
-s)-gff)Settings:
Outputs:
Visualization graphics, including coverage plots, available in the specified output directory (-op).
To view available command-line options and their defaults, use:
toast design -h
toast amplicon_no -h
toast plotting -h
*Decide on SNP priority by modifying the SNP priority file (mutation_priority_example.csv - can be found in github) *Decide on amplicon size
toast amplicon_no -a 800 -op ./cache/Amplicon_design_output -g
toast design -op ./cache/Amplicon_design_output -a 400 -sn 1 -sg rpoB,katG -nn 40
toast design -op ./cache/Amplicon_design_output -a 400 -sn 1 -sg rpoB,katG -nn 25
toast design -op ./cache/output -a 1000 -nn 4 -ud ./cache/test_df.csv
toast design -op ./cache/output -a 1000 -nn 26
toast design -op ./cache/Amplicon_design_output -a 400 -sn 1 -sg rpsL -nn 0 -ud ./cache/test_df.csv
toast plotting -ap ./toast/Amplicon_design_output/Primer_design-accepted_primers-23-400.csv -rp ./toast/db/reference_design.csv -op ./cache/Amplicon_design_output -r 400
user_defined_files/ folder:default_primer_design_setting.txtuser_input_primer.csvSegmented amplicon design is used to generate amplicons of varying sizes so they can be easily distinguished on an agarose gel. This provides a quick visual check to confirm successful amplification before sequencing, allowing users to validate that each target produced a distinct band. It helps avoid wasting sequencing resources on failed reactions and serves as a practical sanity check in the experimental workflow.
If issues arise during amplicon design—such as no primers being generated—it is most likely due to insufficient padding. A small padding size can restrict the available sequence context needed for effective primer placement. To resolve this, try increasing the padding value to provide more room for the design algorithm to work with.
<filetype>-<number of total amplicon designed>-<minimum amplicon size>-<maximum amplicon size>-<step size>-<number of amplicon for each size>
Specific mutation (Mutation Priority)file format: Essentially all you need would be the genome position (genome_pos). Other columns are needed but you could used imputed values like below if unknown. The complete example Mutation priority csv can be found in Github:mutation_priority_example.csv
| sample_id | genome_pos | gene | change | freq | type | sublin | drtype | drugs | weight |
|---|---|---|---|---|---|---|---|---|---|
| sample_1 | 321168 | gene_1 | change_1 | 1 | - | - | - | - | 1 |
| sample_2 | 551767 | gene_2 | change_2 | 1 | - | - | - | - | 1 |
| sample_3 | 1017188 | gene_3 | change_3 | 1 | - | - | - | - | 1 |
| sample_4 | 1119158 | gene_4 | change_4 | 1 | - | - | - | - | 1 |
| sample_5 | 1119347 | gene_5 | change_5 | 1 | - | - | - | - | 1 |
| sample_6 | 1414872 | gene_6 | change_6 | 1 | - | - | - | - | 1 |
You can manually eddit this for though a script (Github:mutation_priority_gen.py) can also be found to generate a file like the above:
example usage:
python mutation_priority_gen.py --positions "322168,553767,1077188" --output <output_path.csv>
REFERENCE: Wang, L., Naphatcha Thawong, Thorpe, J., Higgins, M., Tan, M., Waritta Sawaengdee, Surakameth Mahasirimongkol, Perdigao, J., Campino, S., Clark, T.G. and Phelan, J.E. (2025). A novel tool for designing targeted gene amplicons and an optimised set of primers for high-throughput sequencing in tuberculosis genomic studies. bioRxiv (Cold Spring Harbor Laboratory). doi:https://doi.org/10.1101/2025.01.13.632698.
FAQs
A standalone bacterial amplicon designing tool (Tuberculosis Optimized Amplicon Sequencing Tool)
We found that toast-amplicon demonstrated a healthy version release cadence and project activity because the last version was released less than a year ago. It has 1 open source maintainer collaborating on the project.
Did you know?

Socket for GitHub automatically highlights issues in each pull request and monitors the health of all your open source dependencies. Discover the contents of your packages and block harmful activity before you install or update your dependencies.
Product
Socket’s precomputed reachability slashes false positives by flagging up to 80% of vulnerabilities as irrelevant, with no setup and instant results.

Product
Socket is launching experimental protection for Chrome extensions, scanning for malware and risky permissions to prevent silent supply chain attacks.

Product
Add secure dependency scanning to Claude Desktop with Socket MCP, a one-click extension that keeps your coding conversations safe from malicious packages.