Product
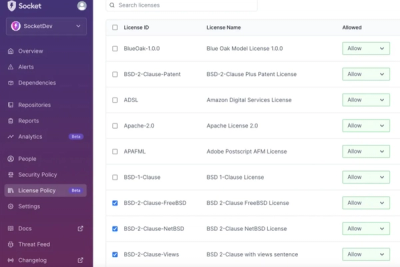
Introducing License Enforcement in Socket
Ensure open-source compliance with Socket’s License Enforcement Beta. Set up your License Policy and secure your software!
By Philipp Burckhardt - Oct 17, 2024
PyGeneBe: A Python client seamlessly integrating with the GeneBe platform, offering efficient annotation of genetic variants through its API, while supporting pandas, VCF file formats, and HGVS parsing
Using this client, you can easily annotate your DNA variants with the GeneBe API. Annotations include:
For more information about the usage, go to the https://pygenebe.readthedocs.io/en/latest/ documentation.
Check current options using --help switch
genebe --help
genebe annotate --help
annotate commandGeneBe client allows you to annotate your VCF file with ease. Use the following command:
genebe annotate --input input.vcf.gz --output output.vcf.gz
Remember that your VCF file must be in a single allelic format! Utilize bcftools (https://samtools.github.io/bcftools/) to split the file. The output VCF will contain additional fields.
To use VCF annoation you have to have cyvcf2 package installed. Take a look at the Installation section below.
If your VCF file is large (over 10.000 variants), you may encounter request limits. To avoid this, create a GeneBe account with an API Key and provide your login/key using the --username and --api-key arguments. You can always check your limits with the account command. Update your annotation command as follows:
genebe annotate --input input.vcf.gz --output output.vcf.gz --username your_username --api-key your_api_key
For more information call
genebe annotate --help
account commandThe account command displays information about your request history statistics and limits. To check your limits without specifying a username and API key, run:
genebe account
Alternatively, if you have a GeneBe account with an API key, use the following command:
genebe account --username your_username --api-key your_api_key
Replace "your_username" and "your_api_key" with your GeneBe account credentials.
For more details and options, you can refer to the help documentation:
genebe account --help
GeneBe makes annotating DNA variants in pandas dataframe easy.
import genebe as gnb
import pandas as pd
input_variants = ['7-69599651-A-G']
# annotate variants with transcripts etc. output as a list; use .netrc for user login and api key
list = gnb.annotate(input_variants,
use_ensembl=True,
use_refseq=False,
genome="hg38",
batch_size=500,
use_netrc=True,
output_format="list")
# output as a pandas dataframe, flat
df = gnb.annotate(input_variants,
use_ensembl=True,
use_refseq=False,
genome="hg38",
flatten_consequences=True,
batch_size=500,
use_netrc=True,
output_format="dataframe")
# parse HGVS, SPDI or other
input_variants_parse = [
"chrX:153803771:1:A",
"22 28695868 AG A",
"22-28695869--G",
"22-28695869-G-",
"NM_000277.2:c.1A>G",
"NM_000277.2:c.2T>C",
"AGT M259T",
"rs1228544607"]
parsed_variants = gnb.parse_variants(input_variants_parse, genome="hg38")
# annotate existing dataframe, using it's chr, pos, ref, alt columns and adding new columns
df = pd.DataFrame({'chr': ['6', '22'], 'pos': [160585140, 28695868], 'ref': ['T', 'AG'], 'alt': ['G', 'A']})
annotated_df = gnb.annotate(df,
genome='hg38',
use_ensembl=False,
use_refseq=True,
flatten_consequences=True,
output_format="dataframe")
# lift over variants from hg19 to hg38
input_variants = ['chr6-161006172-T-G']
from_genome = "hg19"
dest_genome = "hg38"
lifted_variants = gnb.lift_over_variants(input_variants, from_genome, dest_genome)
If you want to annotate thousands of variants, please log in to https://genebe.net, generate an API Key, and provide it using username and api_key or using the .netrc file.
Find out more usage examples in the examples directory.
You can install GeneBe Utils using pip:
pip install genebe
If you wish to install faster mmh3 implementation or use the option of annotating vcf files install using:
pip install genebe[cpp]
or install modules
pip install cyvcf2
pip install mmh3
in the environment.
This step will require build tools installed on your computer.
There is a dockerized version of this package, available at https://hub.docker.com/r/genebe/pygenebe .
Usage example, reading from file input.vcf and writing output to stdout:
docker run -v input.vcf:/tmp/input.vcf --rm genebe/pygenebe:0.0.14 genebe annotate --input /tmp/input.vcf --output /dev/stdout
If you wish to annotate thousands of variants, please log in to https://genebe.net, generate an API Key, and provide it using username and api_key.
The number of daily requests from a single IP is restricted to prevent abuse and excessive resource consumption on our server. Account holders with an API Key enjoy significantly higher limits (in the tens of thousands). If you require a higher daily request limit, please reach out to us via the https://genebe.net .
Experiencing issues? Follow these steps:
Your feedback is crucial for improving GeneBe client. Thank you for contributing to the community!
For more information about GeneBe, visit GeneBe website, https://genebe.net .
FAQs
GeneBe Client: A user-friendly system for annotating genetic variants
We found that genebe demonstrated a healthy version release cadence and project activity because the last version was released less than a year ago. It has 1 open source maintainer collaborating on the project.
Did you know?

Socket for GitHub automatically highlights issues in each pull request and monitors the health of all your open source dependencies. Discover the contents of your packages and block harmful activity before you install or update your dependencies.
Product
Ensure open-source compliance with Socket’s License Enforcement Beta. Set up your License Policy and secure your software!
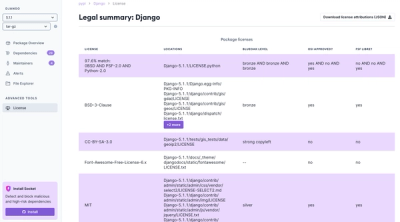
Product
We're launching a new set of license analysis and compliance features for analyzing, managing, and complying with licenses across a range of supported languages and ecosystems.
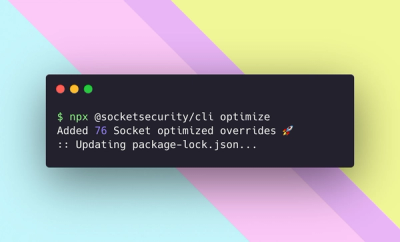
Product
We're excited to introduce Socket Optimize, a powerful CLI command to secure open source dependencies with tested, optimized package overrides.