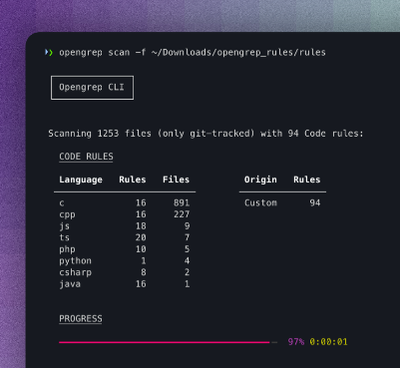
Security News
Opengrep Emerges as Open Source Alternative Amid Semgrep Licensing Controversy
Opengrep forks Semgrep to preserve open source SAST in response to controversial licensing changes.
By Sarah Gooding - Jan 28, 2025
.. image:: https://img.shields.io/github/issues-pr-raw/molssi-seamm/dftbplus_step :target: https://github.com/molssi-seamm/molsystem/pulls :alt: GitHub pull requests
.. image:: https://github.com/molssi-seamm/molsystem/workflows/CI/badge.svg :target: https://github.com/molssi-seamm/molsystem/actions :alt: Build Status
.. image:: https://codecov.io/gh/molssi-seamm/molsystem/branch/master/graph/badge.svg :target: https://codecov.io/gh/molssi-seamm/molsystem :alt: Code Coverage
.. image:: https://github.com/molssi-seamm/molsystem/workflows/CodeQL/badge.svg :target: https://github.com/molssi-seamm/molsystem/security/code-scanning :alt: Code Quality
.. image:: https://github.com/molssi-seamm/molsystem/workflows/Release/badge.svg :target: https://molssi-seamm.github.io/molsystem/index.html :alt: Documentation Status
.. image:: https://img.shields.io/pypi/v/molsystem.svg :target: https://pypi.python.org/pypi/molsystem :alt: PyPi VERSION
Molsystem provides a general class for handling molecular and periodic systems. This is the heart of SEAMM.
This package was created with Cookiecutter_ and the audreyr/cookiecutter-pypackage_ project template.
.. _Cookiecutter: https://github.com/audreyr/cookiecutter
.. _audreyr/cookiecutter-pypackage: https://github.com/audreyr/cookiecutter-pypackage
Developed by the Molecular Sciences Software Institute (MolSSI_),
which receives funding from the National Science Foundation_ under
award OAC-1547580 and CHE-2136142.
.. _MolSSI: https://www.molssi.org
.. _National Science Foundation: https://www.nsf.gov
2025.1.14 -- Bugfix: error reading JSON properties from SDF files * JSON properties were read as strings, not as JSON objects. This is fixed.
2025.1.3 -- Enhanced coordinate precision in SDF files * The full precision coordinates are added as a property to the SDF file. If the full-precision coordinates are aavailable they are used when reading the SDF file.
2024.12.14 -- Bugfix: yet more issues with property handling * This release ensures that types of properties are correctly handled when reading SDF files.
2024.12.14 -- Bugfix: more issues with property handling. * The types of properties were not kept when using Open Babel or RDKit, so when properties were reread from an SDF file the JSON properties were converted into strings, causing various errors. This is fixed.
2024.12.11 -- Bugfix: Properties in SDF files * Transferring properties to the Open Babel and RDKit molecules was incorrect after recent changes to the handling of properties. This fixes the problem, and now SDF files have the properties correctly.
2024.12.7 -- Significant internal enhancement to property handling. * An internal change, allowing listing and getting properties with wildcards, working with multiple values at once. This is a significant change, but should not affect the user interface. Also consolidated the property handling code for configurations vs systems.
2024.11.27.1 -- Added support for charge in chemical formulae * Added support for charge in chemical formulae, e.g. [H2 O]+.
2024.11.27 -- Bugfix: error with charge and multiplicity * The charge and multiplicity of the system were not correctly set when creating a system from a SMILES string using RDKit. More generally, the charge and multiplicity were not correctly set from an RDKit molecule unless explicitly given in the properties.
2024.11.23 -- Bugfix: error if OpenEye not available * Fixed an issue with the import of OpenEye that caused an error if OpenEye was not available.
2024.11.21.1 -- Versions of openbabel, openeye, and rdkit * Added functions to get the versions of openbabel, openeye, and rdkit that are installed.
2024.11.21 -- Added access to OpenEye mols and SMILES * Added the ability to create a system from an OpenEye molecule and vice versa. This gives access to the OpenEye toolkit for generating conformers, etc. * Added the ability to create a system from a SMILES string and create the SMILES string from a system using OpenEye.
2024.11.12 -- Added units to properties in Openbabel molecules * Any properties on the system and configuration are optionally added to the Openbabel Molecule object for e.g. writing to an SDF file. This adds the units of the properties explicitly
2024.10.1 -- Bugfix: Incorrect coordinates from PubChem * Fixed bug where the coordinates from PubChem were accidentally the 2-D rather than 3-D coordinates.
2024.8.17 -- Bugfix: current configuration not updated properly * Existing instances of systems did not correctly update when the default configuration was changed. This is release fixes the problem.
2024.8.5 -- Bugfix: creating H2 from SMILES failed * Fixed bug where creating molecules consisting of just hydrogen failed because RDKit by default ignores all hydrogens when reorienting the molecule.
2024.6.21 -- Switching default for SMILES to RDKit rather than OpenBabel * RDKit seems more robust, and also the atom typing uses RDkit, so compatibility is important.
2024.5.8 -- Added more control over RDKit and OpenBabel creating systems * Added control to from_RDKMol and from_OBMol to allow selectively updating the atoms, coordinates, and bonds
2024.5.6 -- Rotated molecule from SMILES, InChI, or InChIKey to standard orientation * Molecules created from line notation are created in an random orientation. This enhancement rotates them to the standard orientation, which will look nice for small, symmetric molecules.
2024.5.5 -- Bugfix: bonds in RDKit * There was an indexing bug translating bonds back from RDKit to SEAMM. The famous 0/1 problem!
2024.4.6 -- Added gradients * Added gradient on atoms as a separate table alongside atoms, so they take no space unless actually used.
2024.3.13 -- Handle uppercase X, Y, Z in strings for symmetry operators * the Crystallographic Open Database CIF files seems to use upper case X, Y, Z in explicit symmetry operators. These need to be lowercased in the code.
2023.12.5 -- Bugfixes for symmetry * Fixed issue #72, where symmetry was not correctly handled for trigonal and hexagonal cells where atoms had coordinates of 1/3 or 2/3.
2023.11.19 -- Bugfixes in symmetry and CIF files * Reading CIF files could fail if the symmetry operators were given * The symmetry handling did not recognize hexagonal spacegroups without :H. Changed so if the hexagonal group name has neither :H or :R, the hexagonal setting is assumed. * When finding the spacegroup from the symmetry operators, hard-coded to the P1 case to avoid what seems like a bug in spglib. * Enhanced to use the full International Tables HM name for spacegroups, translating the input to that standard name.
2023.11.5 -- Bugfix and improved symmetry handling * Fixed bug with symmetry operators containing blanks, e.g. 'x, y, z' rather than 'x,y,z' * Added handling of symmetry when get properties of atoms * Added method to lower symmetry to P1/C1
2023.10.30 -- Support for InChI improved, RMSD and PubChem added... * Adds support for aligning structures and calculating RMSD * Adds support for working directly with PubChem to get structures, IUPAC names, etc. * Improves support for InChI, working around issues in both OpenBabel and RDKit. * Added substantial new functionality for spacegroups and primitive cell handling, but still not complete.
2023.9.20 -- Better support for primitive cells and spacegroups * Added getting the spacegroup from the symmetry operators * Fixed updating the coordinates from the primitive cell
2023.9.5 -- Support for velocities of atoms.
2023.8.30 -- Support for spacegroup symmetry.
2023.8.27 -- Bugfix: writing SDF did not handle charge and multiplicity.
2023.7.30 -- Improved handling of properties * Added ability to get lists of systems or configurations filtered by name * Improved handling of properties on just a system, not configuration * Added ability to filter properties retrieved * Improved handling of properties when creating OpenBabel OB_MOL object
2023.7.26 -- Bugfix: error in QCSchema bonds; enhancement: RDKit * Fixed bug in the bond indices in QCSchema * Added ability to use RDKit for SMILES and InChI
2023.7.18.1 -- Added support for creating structures from InChIKeys * Uses PubChem to translate the InChiKey to InChI.
2023.7.18 -- Added support for InChI and InChIKeys
2023.7.9 -- Added JSON properties * Added properties stored as JSON, which allows, vectors, tensors, etc.
2023.4.6 -- Enhancements for CIF files * Handle uncertainties in CIF files expressed as '(x)' at end of value.
2023.3.30 -- Enhancements to QCSchema support * Improved naming of molecule in QCSchema * Added creation of configurations from QCSchema objects.
2023.2.13 -- Fixed issue with valence in RDkit for cations like NH4+
2022.11.20 -- Added a method to copy a configuration.
Added a new method to the system class, copy_configuration, that creates a copy of
the configuration using the same atomset and bonset, but new coordinates and cell so
that any changes to coordinates and cell are not shared between the configurations. By
default it copies the current configuration.
2022.11.18 -- Fixed bug with handling for Open Babel The total charge and multiplicity were not correctly set when creating an Open Babel molecule.
2022.11.3 -- Add handling of strain and improved handling of properties Added methods for straining the unit cell, and also straining a configuration, correctly handling the coordinates for an affine transformation. In the future will add e.g. affine transformation of the centers of molecules, which is useful for molecular fluids.
Added the system for properties, in addition to the configuration. This allows system properties that are not associated with a particular configuration, which is often appropriate for experimental results. It also makes it much easier to search for systems where any configuration has a particular property.
2022.10.26 -- Improved database write performance. Switched to write-ahead mode and tweaked memory settings. This gives a large performance improvement (10x or more) for large database (~1 GB).
FAQs
molsystem
We found that molsystem demonstrated a healthy version release cadence and project activity because the last version was released less than a year ago. It has 2 open source maintainers collaborating on the project.
Did you know?

Socket for GitHub automatically highlights issues in each pull request and monitors the health of all your open source dependencies. Discover the contents of your packages and block harmful activity before you install or update your dependencies.
Security News
Opengrep forks Semgrep to preserve open source SAST in response to controversial licensing changes.

Security News
Critics call the Node.js EOL CVE a misuse of the system, sparking debate over CVE standards and the growing noise in vulnerability databases.

Security News
cURL and Go security teams are publicly rejecting CVSS as flawed for assessing vulnerabilities and are calling for more accurate, context-aware approaches.